1. Organic thermoelectric materials
Thermoelectric materials can inter-convert heat and electricity utilizing the Seebeck and Peltier effects, which have great potential for deep space applications and waste heat harvesting. Conventional thermoelectric materials are inorganic semiconductors with narrow band gaps, such as Bi2Te3. With the success of organic electronics, organic semiconductors that are flexible, easy to process, and low-cost are drawing attention for thermoelectric applications.
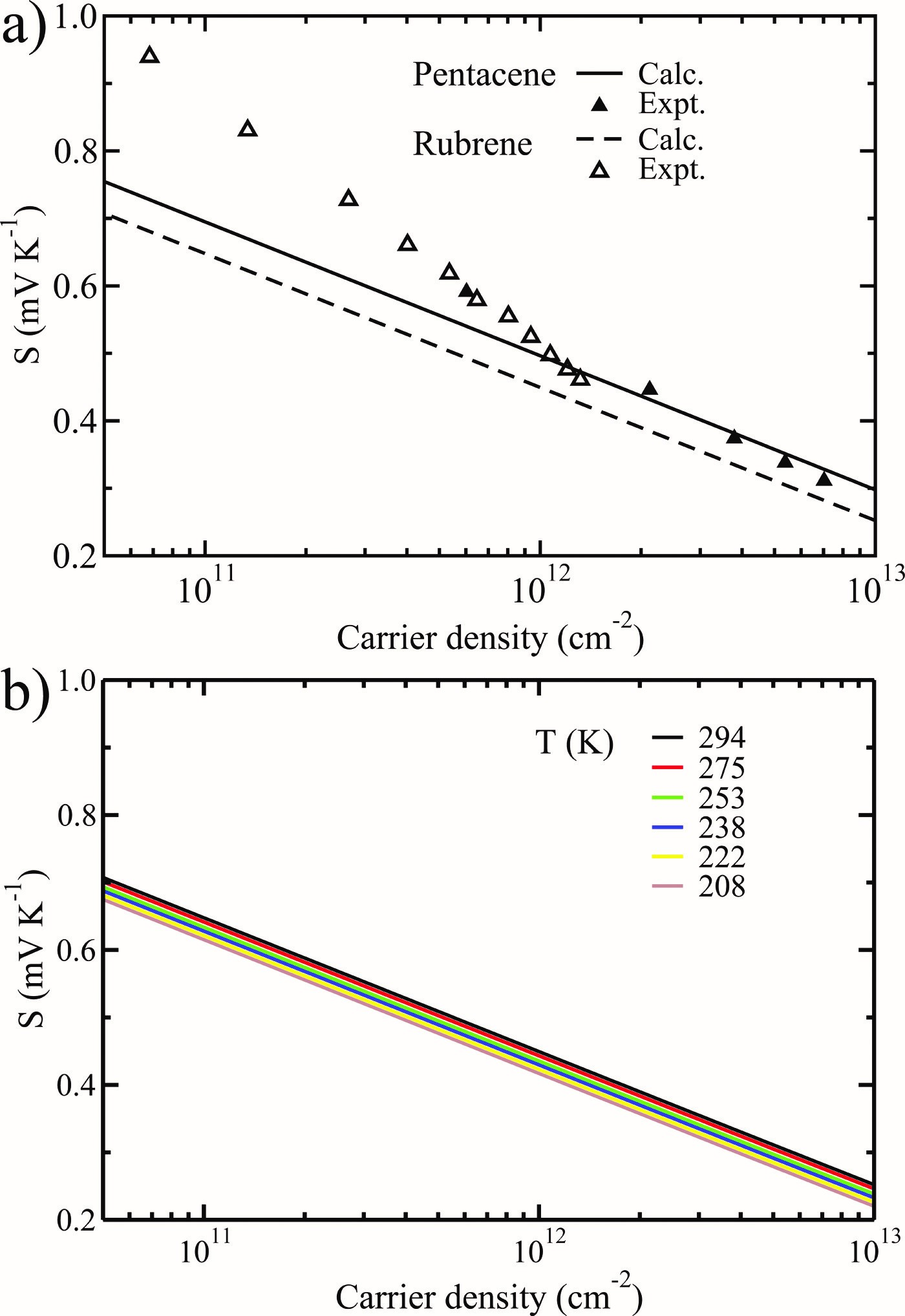
(1) We started research on organic thermoelectric materials in 2009, with two seminal works on thermoelectric transport in pentacene and rubrene within the framework of Boltzmann transport theory (J. Chem. Phys. 2009, 131, 224704), and lattice thermal transport based on the non-equilibrium molecular dynamics simulation (J. Phys. Chem. C 2011, 115, 5940-5946). The agreement between theory and experiment is satisfying, which gives us confidence to design organic thermoelectric materials on a theoretical basis.
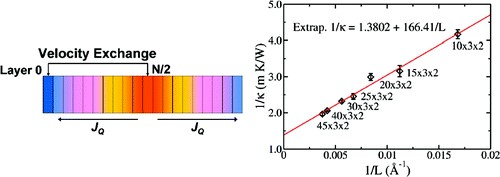
(2) In order to solve the Boltzmann transport equation (BTE), the relaxation time approximation is usually adopted. In the 2009 paper, we took a constant empirical parameter as the relaxation time of charge carriers. In fact, obtaining charge carrier relaxation times in organic semiconductors from first-principles is really challenging and computationally demanding. We first applied the deformation potential theory proposed by Bardeen and Shockley in 1950 to model electron-acoustic phonon interactions in phthalocyanines (Pcs) and benzothienobenzothiophenes (BTBTs), see J. Chem. Theory Comput. 2012, 8, 3338-3347 and Chem. Mater. 2014, 26, 2669-2677.
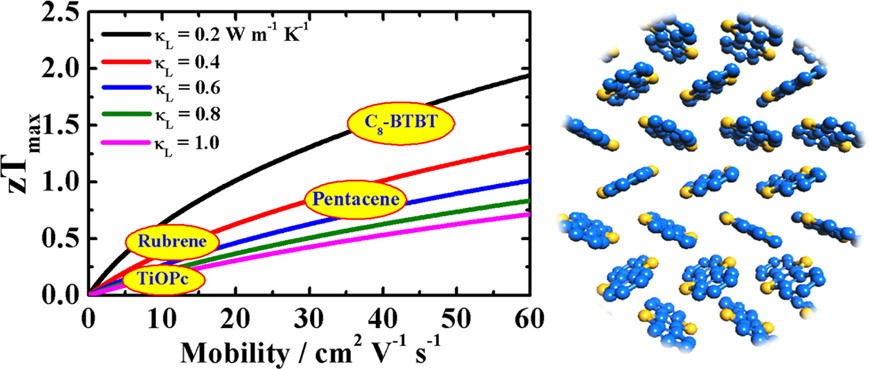
(3) Despite of the simplicity of the deformation potential theory, the predicted mobility of C12-BTBT and Seebeck coefficient of C8-BTBT have been verified respectively by the experiments of Seki’s group at Kyoto University and Sirringhaus’ group at Cambridge University (see their papers published in Adv. Mater. 2016, 28, 7106 and APL Mater. 2015, 3, 096104). The combination of deformation potential theory with BTE thus provides a parameter-free scheme to model thermoelectric transport in non-polar crystals, which has become very popular in predicting charge transport and thermoelectric properties of emerging materials.
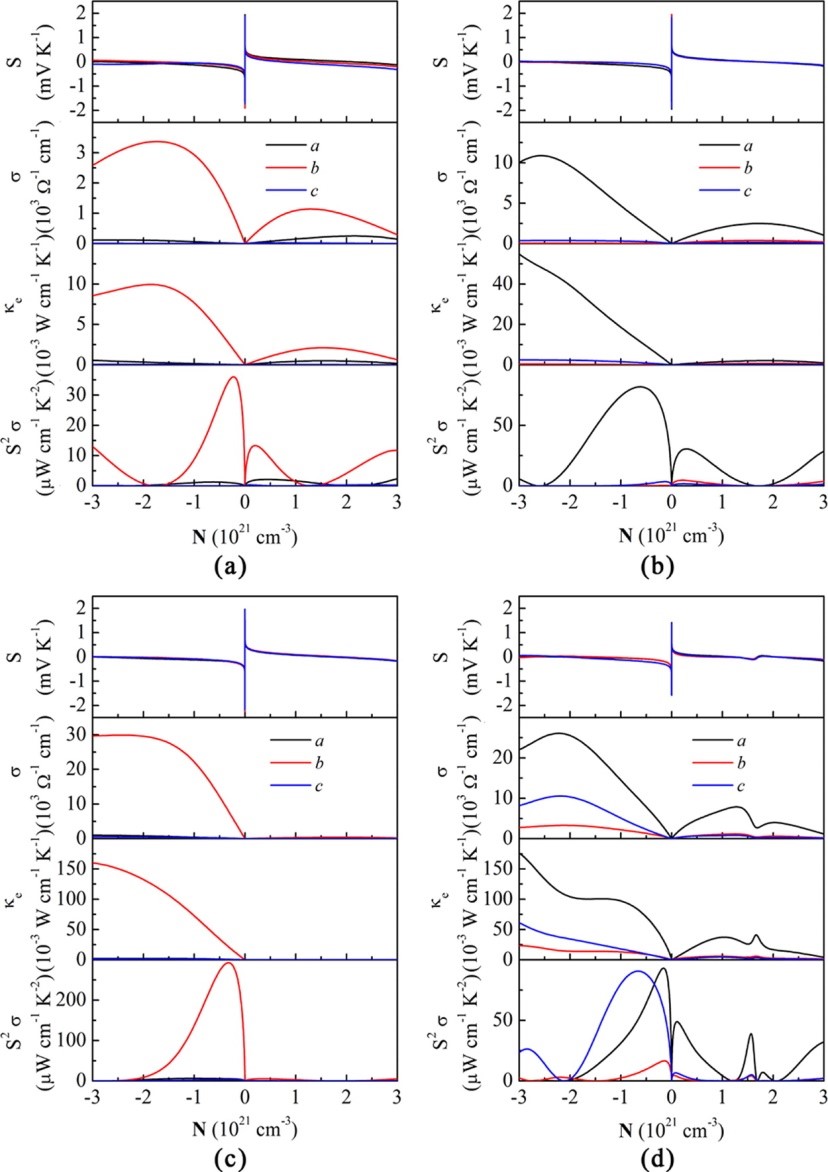
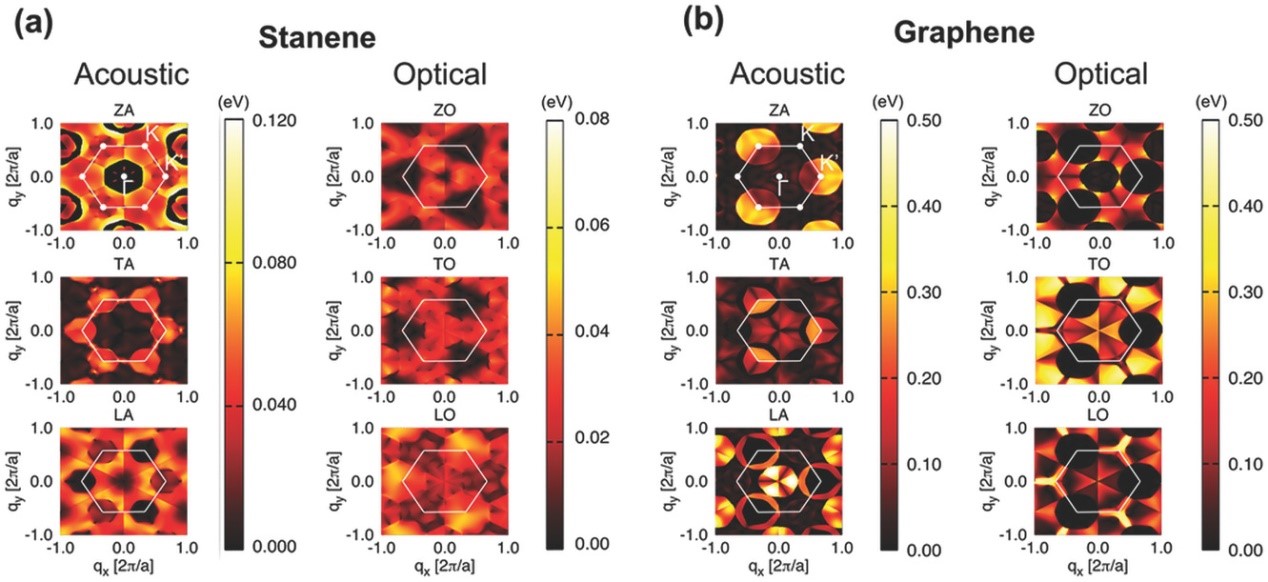
(4) In non-polar crystals, acoustic phonon scatterings play a dominant role, yet omission of optical phonon scatterings will certainly lead to the overestimation of charge carrier mobility. We are now making an effort to derive all the electron-phonon interactions based on density functional perturbation theory (DFPT) and Wannier interpolation method. Applications to monolayer materials such as stanene (Adv. Electron. Mater. 2017, 3, 1700143) and GeAs2 (Chem. Mater. 2017, 29, 6261-6268) have been realized, and application of the new method to organic materials is an on-going project.
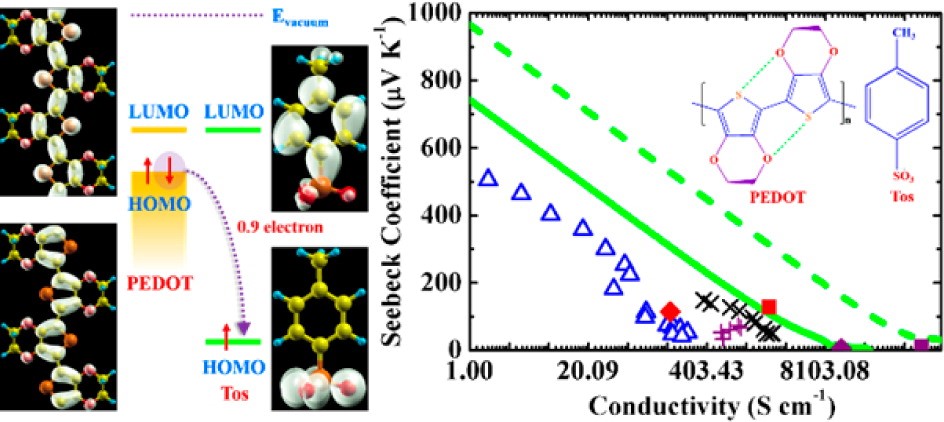
(5) In addition to developing reliable computational methods for charge carrier scatterings, an important issue in developing organic thermoelectric materials is the role of chemical doping. Redox doping introduces charge carriers to organic semiconductors. The charges first fill the trap states resulting from impurities or defects, and the remaining charge carriers are injected into the delocalized electronic states, which will significantly improve the performance of organic electronic devices. In the case of thermoelectric conversion, an accurate control of doping level is of utmost importance, since the electrical conductivity increases while the Seebeck coefficient decreases rapidly with the carrier concentration, giving rise to the maximum power factor at a certain carrier concentration. We demonstrate that doping not only increases the carrier concentration, but also introduces an ionized scattering center to charge carriers. We model the ionized impurity scattering by screened Coulomb potential of Brooks-Herring, and show that such a scattering mechanism dominates over the acoustic phonon scattering in PEDOT:Tos, a p-doped thermoelectric polymer with high figure of merit (J. Am. Chem. Soc. 2015, 137, 12929-12938).
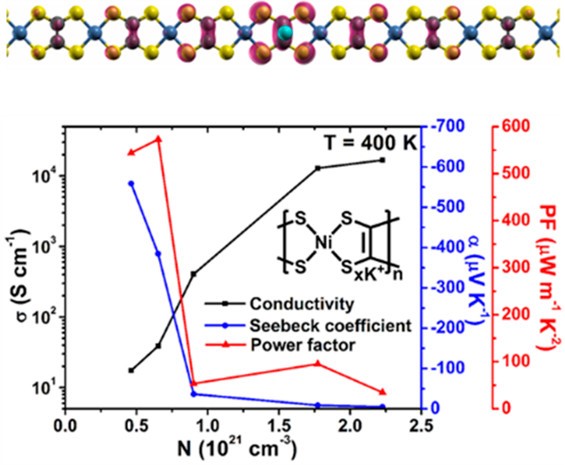
(6) In K-doped poly[Ni-ett], an n-type metal coordination polymer, we observed doping induced polaron band formation, which boosts the Seebeck coefficient and power factor at elevated temperatures due to the thermal activation of electrons from the half-filled polaron band to the conduction band (J. Phys. Chem. Lett. 2019, 10, 2493-2499). Our two-band transport model explained the non-monotonic temperature dependence of the Seebeck coefficient observed in the experiment, and predicts that such an unusual behavior can also be observed in other polymers under appropriate doping conditions.
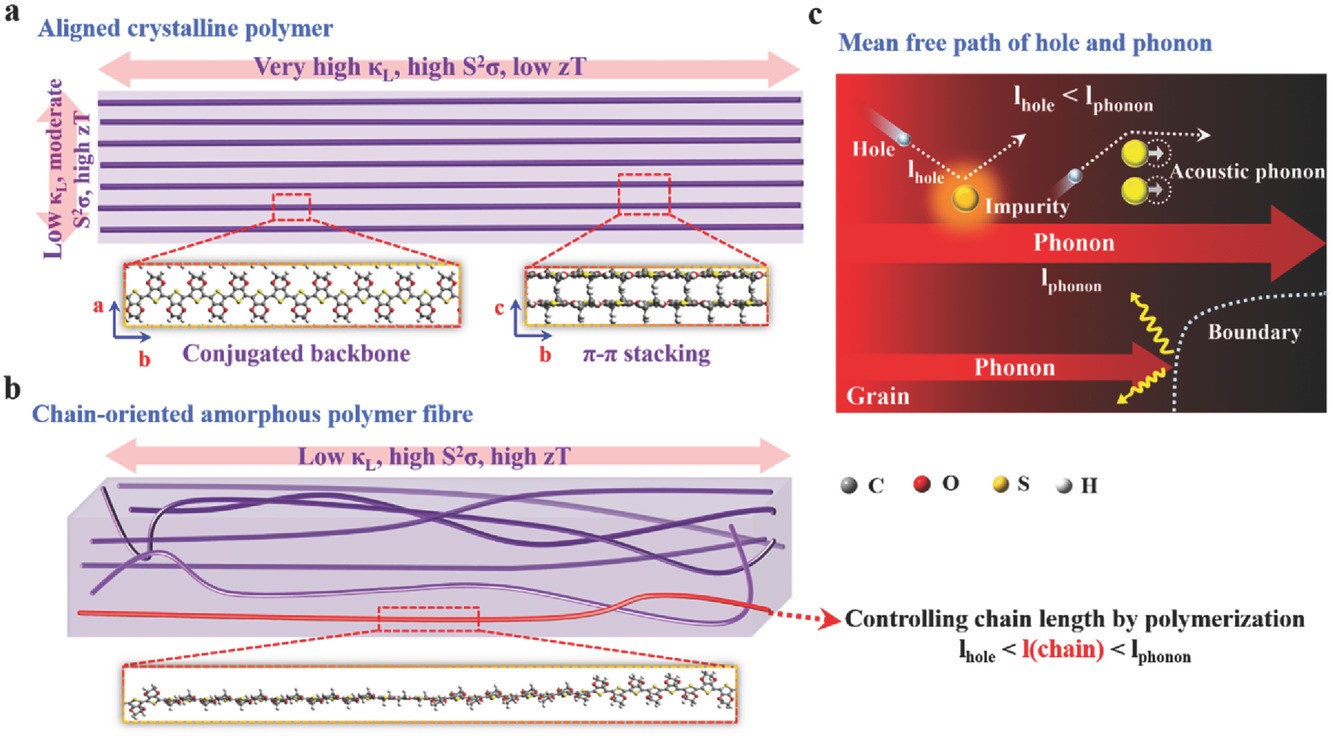
(7) High-performance thermoelectric materials should be both electron-crystal and phonon-glass for improved figure of merit. Using molecular dynamics simulations, we manifest that such a criterion can be achieved in chain-oriented conducting polymers by regulating the degree of polymerization and the crystallinity of polymers (Adv. Funct. Mater. 2017, 27, 1702847). The heat carriers, i.e., phonons travel along the polymer backbone over a distance much longer than the charge carriers do before they are scattered, so when a polymer chain is shorter than the mean free path of phonons, yet longer than that of charge carriers, the phonon transport along the polymer chain will be suppressed to a large extent while the charge transport will be barely affected. The strategy we proposed to engineer thermal transport provides an alternative route for achieving high thermoelectric efficiency other than band engineering and doping engineering.
The theory and computational methods to model thermoelectric transport in organic materials, as well as theoretical insight and design strategies for high-performance organic thermoelectric materials are summarized in our two perspective reviews (Phys. Chem. Chem. Phys. 2012, 14, 16505-16520; Adv. Electron. Mater. 2019, 5, 1800882).

2. Design of COFs and MOFs for spintronic, magnetic and photocatalytic applications
Covalent organic frameworks (COFs) and metal organic frameworks (MOFs) are two emerging classes of organic and organic-inorganic hybrid porous materials with high designability and chemical diversity. Their intriguing properties including ultrahigh porosity, tunable functionality, and low density make them promising for gas storage and separation, catalysis, light-harvesting and energy conversions. Our focus is on the magnetic properties of COFs and MOFs.
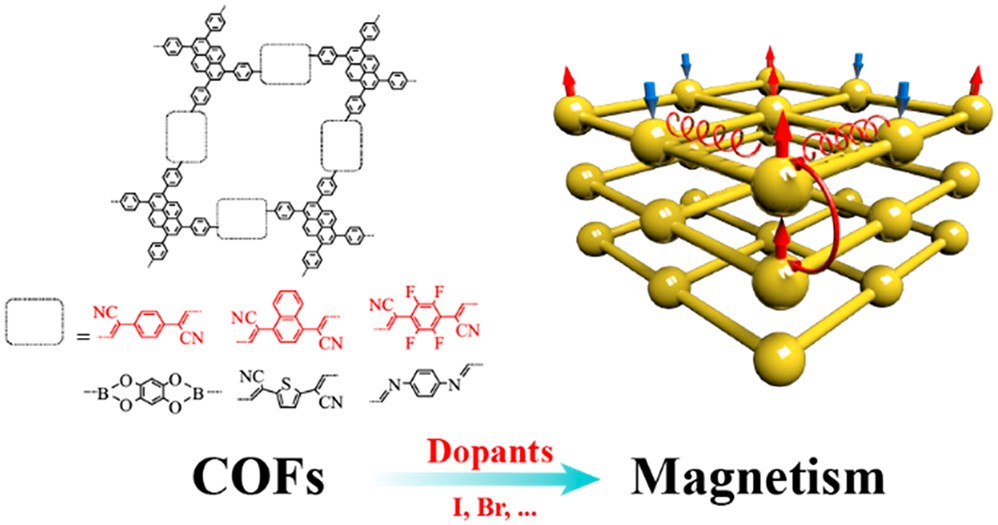
Organic and molecule-based magnets whose unpaired electron spins reside in p-orbitals are a new and growing class of magnetic materials exhibiting ferromagnetic or other types of magnetic behavior. However, stable organic materials often lack unpaired electrons. We proposed a general approach to attain unpaired electron spin and metal-free magnetism in narrow-band COFs by chemical doping (J. Am. Chem. Soc. 2020, 142, 11013-11021). We found that dopants with energy-matched frontier orbitals to COFs not only inject charges, but also further localize the charges through orbital hybridization and the formation of a supramolecular charge-transfer complex. We designed two new COFs with narrow valence bands, which show prospective magnetism after doping with iodine, and demonstrated that both spin-conduction and magnetic interactions can be effectively modulated by manipulating the chemical building blocks of COFs
3. 2D materials for thermoelectric conversion, ferroelectric and structural phase transition memories
A large variety of 2D and quasi-2D materials have emerged since the successful exfoliation of graphene in 2004. These materials call attention not only for their unique and extraordinary properties arising from the size and quantum confinement effect, but also for the ease to be integrated into nanodevices.
(1) We investigated thermoelectric and thermal transport properties of group IV-V compounds GeAs2, group-V arsenene and arsenic-phosphorene from first-principles. Ge and As fall within the same period as selenium in periodic table, and GeAs2 exhibits anisotropic and anharmonic bonding similar to the IV−VI group compound SnSe, which is a promising thermoelectric material with layered crystal structure. We uncover amazingly low lattice thermal conductivity in the monolayer of GeAs2, which can be attributed to its soft vibrational modes and anomalously high Grüneisen parameter. The ultralow thermal conductivity makes GeAs2 promising for environmentally friendly thermoelectric applications (Chem. Mater. 2017, 29, 6261-6268).
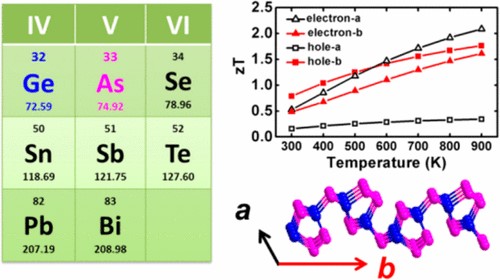
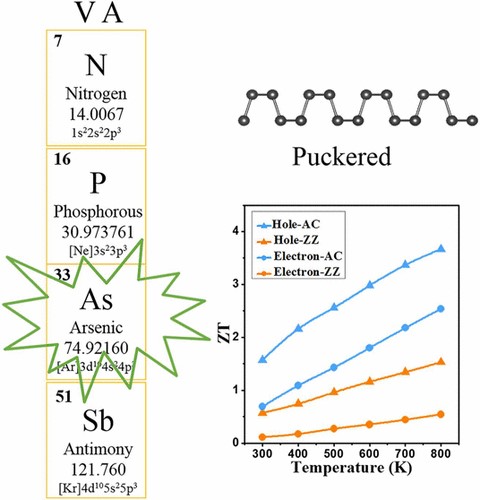
(2) The group-V monolayers of puckered structure exhibit exotic anisotropy of transport: the preferential direction of charge transport is orthogonal to the preferential direction of lattice thermal transport, which can benefit the thermoelectric conversion in one direction and suppress that in the other (J. Phys. Chem. C 2017, 121, 19080-19086 and Phys. Chem. Chem. Phys. 2018, 20, 14024-14030).
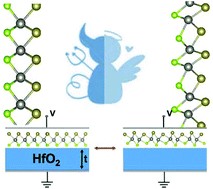
(3) Phase transition materials are widely exploited in sensors, switches, and information storage devices. The reversible semiconductor-to-semimetal structural phase transition in the monolayer of MoTe2 by the electrostatic gating has been recently reported. To discover more 2D phase transition materials, we screen in the Janus family of transition metal dichalcogenides, MXY, where M = Mo or W, X/Y = S, Se, or Te. We find that the Janus monolayer of WSeTe can undergo reversible phase transition modulated by the electrostatic gating, owing to the small energy difference between its 2H and 1T’ phases. The kinetic barrier for phase transitions in WSeTe is significantly lower than that in MoTe2, which can cause three orders of magnitude increase in the transition rate and more rapid response of devices (Nanoscale 2018, 10, 21629).
(4) Ferroelectric materials are materials that can exhibit spontaneous electrical polarizations and the direction of the polarization can be reversed by applying an external electric field. They have been widely applied in non-volatile memories, field-effect transistors, sensors and other devices. Van der Waals layered materials are lack of dangling bonds when exfoliated into thin layers, so they are ideal candidates for ultra-thin ferroelectric applications. So far, out-of-plane polarizations in 2D materials are only reported for distorted 1T-MoTe2 and α-In2Se3 monolayers. We predict that two metal phosphorous trichalcogenides (MPTs), CuInP2S6 and CuBiP2S6, exhibit the above room-temperature ferroelectricity down to the single-layer limit. The polarizations can persist in van der Waals heterostructures. Both band gaps and band alignment of such ferroelectric heterostructures can be further modulated by reversing the vertical polarization with an electric field. The heterostructure of CuInP2S6/Zn2P2Se6 with band gap and type-II band alignment suitable for photocatalytic water splitting is demonstrated.
4. Examples of experimental collaborations
We have close collaborations with experimentalists on various chemical problems, including surface and solution self-assembly, electrocatalysis and photocatalysis. To solve these problems, knowledge of various intermolecular interactions and their cooperation and competition, electronic structure and properties of catalysts, catalytic reaction mechanism and free energy change is required, which can be only learned from theoretical modelling and computations. Through collaborations, we aim to provide the molecular picture for experimental observations, discover the underlying mechanism, guide the molecular design of materials, and ultimately to realize our predictions in the lab. A few examples of experimental collaboration are given below.
(1) Our collaborators found that oleamide, a long-chain alkene with cis-C=C bond and amide terminal group, self-assembles into 1D nanoribbons along the zigzag direction of MoS2. Based on the hydrogen bonding interactions of oleamide and van der Waals interactions between oleamide and MoS2, we proposed a molecular assembly model which gave rise to the oriented assembly exactly along the zigzag direction of MoS2 (Nat. Commun. 2017, 8, 377).
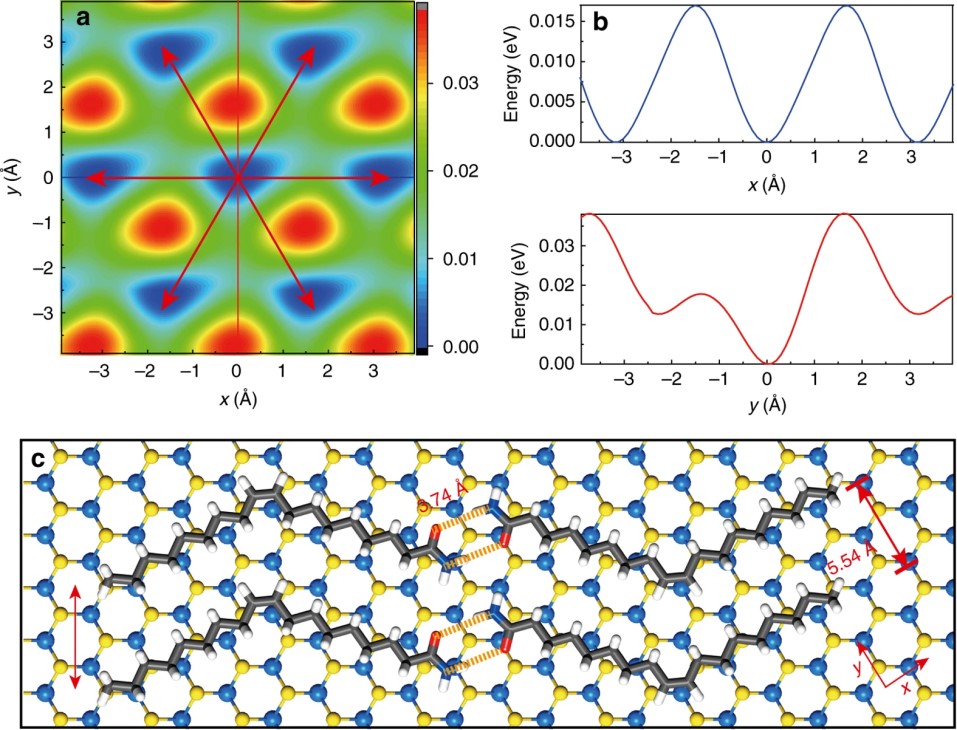
(2) Further, we found that either cis-C=C bond or amide group is required for the oriented assembly on both hexagonal and orthorhombic atomic lattices. Our prediction on the assembly of other long-chain cis-alkene and derivatives on graphene and black phosphorene was also verified by the experiment (J. Phys. Chem. C 2019, 123, 30996-31002).
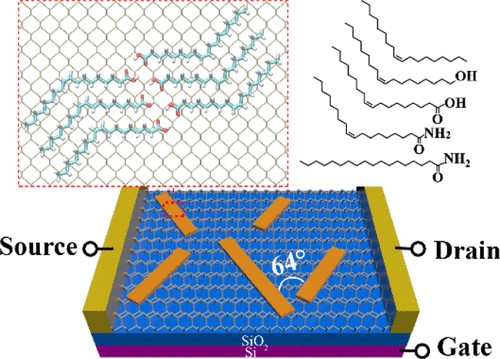
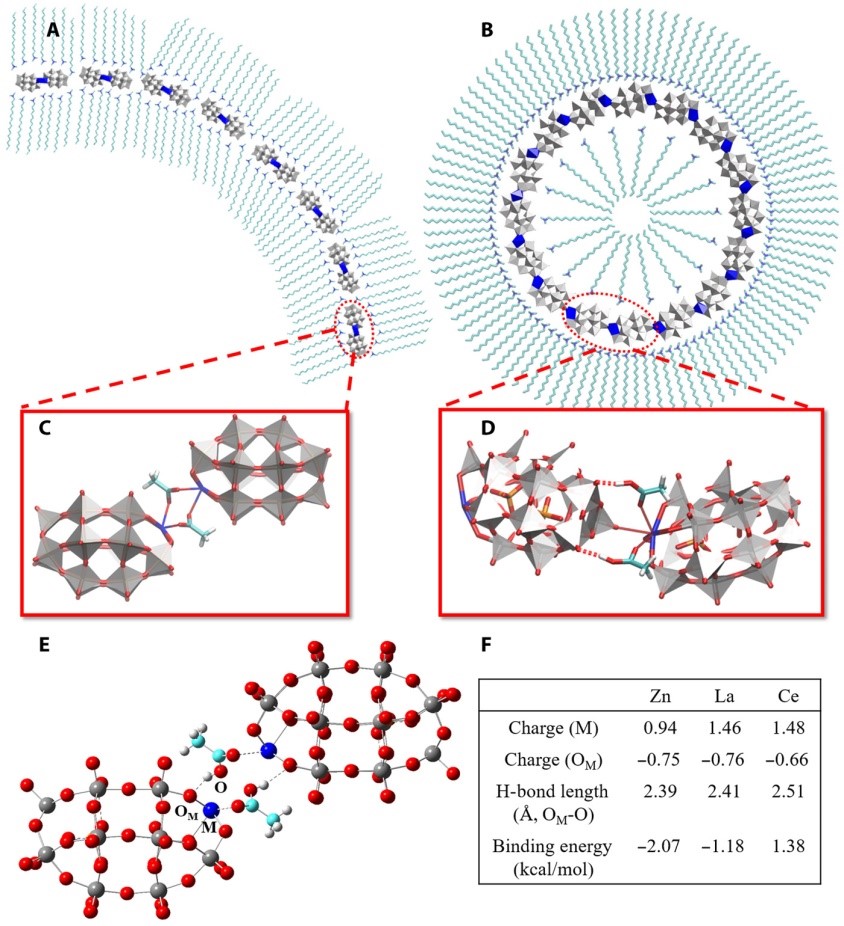
(3) Polyoxometalates (POMs) are sub-nanometer-sized clusters with a variety of structures. Our collaborators constructed a series of single-cluster nanowires, single-cluster nanorings, and three-dimensional superstructure assemblies by single POM clusters. We identify that HAc can mediate the assembly of POM clusters, and it is responsible for the pH-regulated conversion from nanowires to nanorings. Based on potential intermolecular interactions between POM, HAc/Ac-, and surfactants as well as the first-principles calculations, we proposed a molecular model for nanorings and nanowires, in which the assembly of POMs is mediated by HAc and Ac-, respectively. When the pH of the solution is higher than the pKa (4.5) of HAc POMs assemble into nanowires, and when the pH is 4.0 they assemble into nanorings (Sci. Adv. 2019, 5, eaax1081).
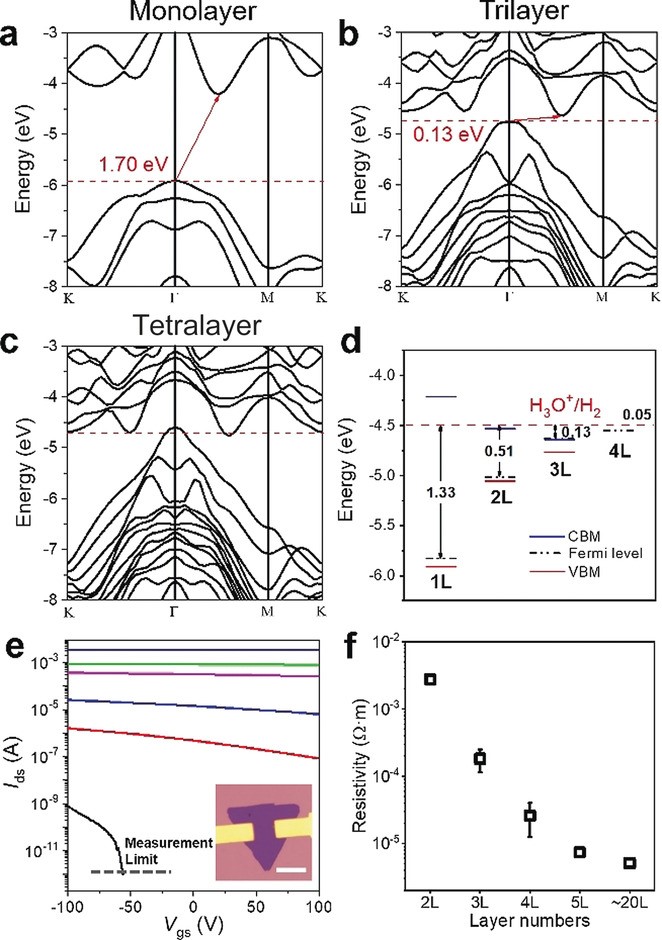
(4) PtSe2 shows the most prominent layer‐dependent electrical properties among various 2D materials and high catalytic activity for hydrogen evolution reaction (HER). We investigated the layer‐dependent HER catalytic activity of 2D PtSe2 from both theoretical and experimental perspectives. Our calculations show that the metal-semiconductor transition occurs between tetralayer and trilayer: PtSe2 of monolayer, bilayer, and trilayer is indirect band gap semiconductor, and that of tetralayer and above is metal. We correlate the improved HER performance of thicker PtSe2 to the improved conductivity due to the semiconductor-metal transition, and the more efficient heterogeneous electron transfer from PtSe2 to H+, as manifested by the smaller energy difference between the Fermi level of few-layer PtSe2 and the absolute electrical potential of standard hydrogen electrode. The unsaturated Pt or Se site at the edge of PtSe2 flakes is identified as the catalytic active site for HER (Angew. Chem. 2019, 58, 6977-6981).
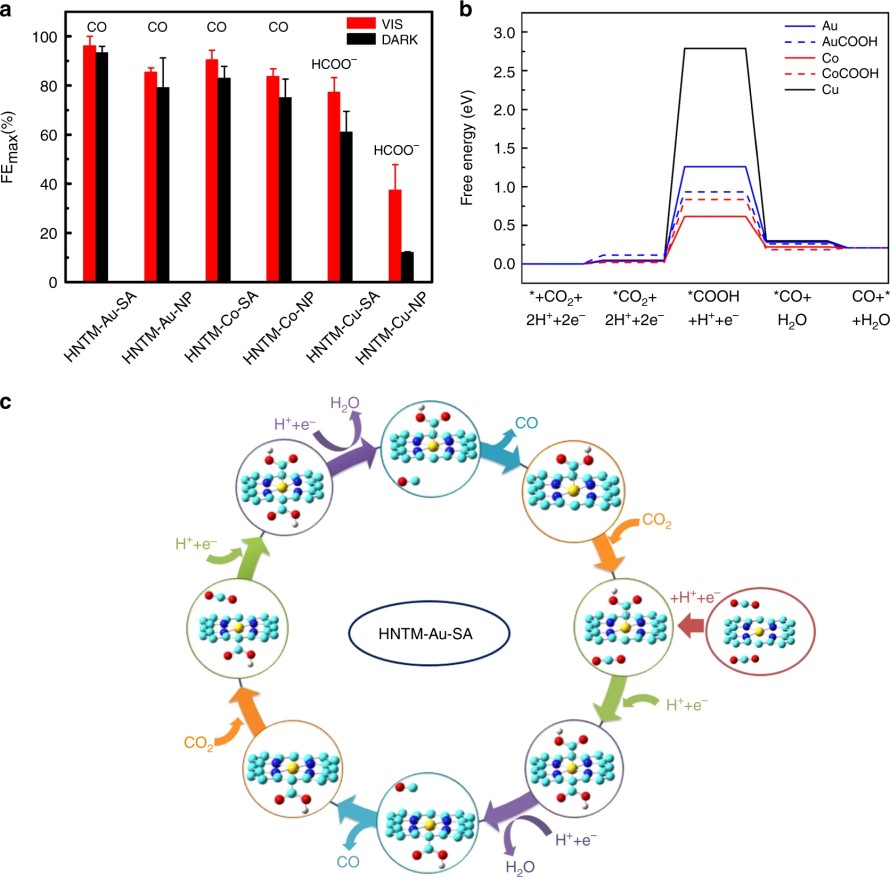
(5) In an experimental collaboration to understand photo-coupled electroreduction of CO2, we calculated the free energy landscape for CO2 reduction on metal porphyrin catalysts with the computational hydrogen electrode model. The conversion of CO2 to CO is a two-electron two-proton process. We found that the first proton-coupled electron transfer step, in which the adsorbed *CO2 is transformed to *COOH is rate-determining, and it is endothermal with the free energy change the lowest on Co porphyrin catalyst, higher on Au-porphyrin, and the highest on Cu porphyrin catalyst. The metal atom in metal porphyrin catalysts is coordinated by four N atoms, and it can adsorb two CO2 simultaneously. We proposed a synergistic catalysis mechanism for Au porphyrin catalyst, which shows that with one CO2 adsorbed onto the catalyst from one side of the porphyrin plane, the free energy change of the rate-determining step on the other side is lowered. Such synergistic mechanism is not observed for Co porphyrin, which is in corroboration with the experimental observation that Au porphyrin catalyst exhibited a much higher turnover frequency than Co porphyrin catalyst (Nat. Commun. 2019, 10, 3844).